ANGEITIS Y GRANULOMATOSIS PULMONARES
En 1973 Liebow introdujo el término de angeítis y granulomatosis pulmonares para definir un grupo de lesiones caracterizadas por necrosis tisular acompañada de reacción granulomatosa y vasculitis. Bajo este epígrafe se reunieron varios procesos (Tabla 6) que presentaban entre sí sustanciales diferencias clínicas, histológicas y patogénicas. Pero, ciertamente, este concepto ayudó mucho a entender una serie de entidades que compartían un patrón morfológico caracterizado por infiltración celular de las paredes de los vasos (angeítis) y cambios necróticos y organizativos del parénquima pulmonar (granulomatosis). La tabla 7 resume las características diferenciales más importantes entre estas entidades.
TABLA 6.-Clasificación de las Angeitis y Granulomatosis Pulmonares
| Granulomatosis de Wegener Angeitis granulomatosa y alérgica de Churg-Strauss Granulomatosis linfomatoide Angeitis y granulomatosis linfocítica benigna Granulomatosis necrotizante sarcoidea y Vasculitis granulomatosa asociada a Sarcoidosis Granulomatosis broncocéntrica Vasculitis granulomatosa asociada a granulomas infecciosos (TB, hongos, etc.) |
TABLA 7.- Diagnóstico diferencial de las Angeitis y Granulomatosis pulmonares.
| GW | GACS | GNS | NEC | GBC | INF | |
|---|---|---|---|---|---|---|
| ASMA | – | ++ | – | ++ | + | – |
| EOSINOFILIA PERIFÉRICA | – | ++ | – | + | + | – |
| ENF. EXTRAPULMONAR | + | + | – | – | – | +/- |
| EOSINOFILIA PULMONAR | +/- | ++ | – | ++ | + | – |
| GRANULOMAS SARCOIDEOS | raros | raros | ++ | +/- | +/- | + |
| GRANULOMAS BRONQUIALES | + | + | + | – | ++ | + |
| MICROORGANISMOS | – | – | – | – | + | + |
GW: Granulomatosis de Wegener; GACS: Granulomatosis alérgica de Churg-Strauss; GNS: Granulomatosis necrotizante sarcoidea; NEC: Neumonía eosinófila crónica; GBC: anulomatosis broncocéntrica; INF: granulomas infecciosos.
Granulomatosis de Wegener
La Granulomatosis de Wegener (GW) es una enfermedad de etiología desconocida que se caracteriza por la presencia de vasculitis granulomatosa necrotizante del aparato respiratorio superior e inferior, glomerulonefritis y vasculitis de pequeño vaso de variable distribución. Es poco frecuente y su incidencia real no se conoce con exactitud, pero se estima que es de 0’4/100.000 habitantes. En la mayoría de los casos, se manifiesta en la cuarta o quinta década de la vida. Predomina ligeramente en el sexo masculino, con una proporción de dos varones por mujer.
La GW se presenta generalmente con síntomas referidos a la vía aérea superior, en forma de sinusitis, rinorrea, obstrucción nasal, afonía, gingivitis y deformación del tabique nasal en silla de montar. Otros síntomas de presentación son: cefalea, sindrome constitucional, artromialgias y lesiones cutáneas. El pulmón es el órgano que se afecta con más frecuencia en la GW evolucionada, manifestándose como tos, disnea, hemoptisis o dolor torácico. La radiología de tórax muestra la presencia de nódulos pulmonares sin niguna localización típica, que tienden a la cavitación y que pueden ser transitorios (figura 5). La afección renal es el requisito indispensable para la GW evolucionada. Se presenta con alteraciones del sedimento urinario, principalmente hematuria, proteinuria y cilindruria. Posteriormente puede evolucionar a insuficiencia renal terminal. La frecuencia de afección de otros órganos se muestra en la tabla 8.
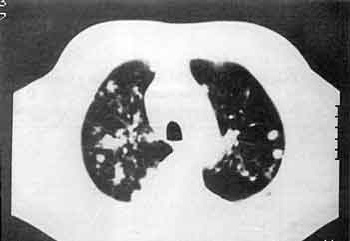
Figura 5.- Imagen de TAC de nódulos pulmonares multiples y periféricos en un paciente de 73 años diagnosticado de Granulomatosis de Wegener.
TABLA 8.- Frecuencia de afectación de distintos órganos en la GW
| ÓRGANO | % |
|---|---|
| Pulmón | 100 |
| Senos paranasales | 95 |
| Nasofaringe | 91 |
| Tracto urinario | 81 |
| Articulaciones | 57 |
| Piel | 48 |
| Ojos | 43 |
| Oídos | 38 |
| Corazón | 29 |
| Sistema nervioso | 24 |
Una de las características más singulares de la GW, que durante los últimos años ha conseguido despertar un creciente interés, es la presencia de un marcador serológico dé indudable utilidad en el diagnóstico: los anticuerpos anticitoplasma del neutrófilo (ANCA), que se han diferenciado básicamente en dos tipos, perinuclear (P-ANCA) y citoplasmático (C-ANCA). En la GW, más del 80% de los pacientes tiene ANCA, de los cuales casi el 95% son del tipo C-ANCA y el resto P-ANCA. La frecuencia se reduce considerablemente en pacientes con poliarteritis nudosa y síndrome de Churg-Strauss: Más notable es el porcentaje de ANCA en pacientes con glomerulonefritis necrosante, con o sin vasculitis sistémica acompañante, ya que se acerca al 83% y de éstos, el 80% pertenecen al tipo P-ANCA mientras el 20% restante son C-ANCA, lo que contrasta con la GW.
El diagnóstico se establece de forma definitiva por la demostración de una vasculitis granulomatosa en un contexto clínico y analítico (ANCA) compatible. La biopsia pulmonar por minitoracotomía o videotoracoscopia es el procedimiento que ofrece una mayor rentabilidad.
Pero el cambio posiblemente más sustancial que se ha producido en la GW es, sin duda, en su pronóstico porque se dispone en la actualidad de un tratamiento que ha demostrado su eficacia en un porcentaje no despreciable de enfermos. y más, si se tiene en cuenta que apenas hace unas décadas la mortalidad de la GW superaba el 90% a los dos años del diagnóstico, con una supervivencia media, sin tratamiento, de dos meses. Con la ciclofosfamida, sola o con corticoides, se ha conseguido la remisión prolongada en más del 90% de los enfermos y hasta, incluso, en algunos su curación. Aún se desconoce el papel concreto que juega el cotrimoxazol en el tratamiento de la GW, pero su utilización se ha justificado al observarse que frecuentemente las recaídas van precedidas por alguna infección. Para Specks y DeRemee, el cotrimoxazol estaría indicado en las formas limitadas, como complemento al tratamiento convencional de la GW generalizada y/o de manera preventiva en las fases de remisión.
Angeítis Granulomatosa y alérgica de Churg-Strauss.
La angeítis granulomatosa y alérgica de Churg-Strauss (GACS) es un síndrome clínico caracterizado por una vasculitis sistémica asociada a eosinofilia periférica, que acontece casi exclusivamente en individuos con asma y rinitis alérgica.
Clínicamente, la fase prodrómica está constituida por la enfermedad alérgica, asma bronquial y/o rinitis. Esta fase precede habitualmente durante meses o años a la fase de vasculitis; más raramente, aparecen de manera simultánea. La segunda fase se caracteriza por la eosinofilia sanguínea e hística y produce cuadros indistinguibles del síndrome de Loeffler, de la neumonia eosinófila crónica o de la gastroenteritis eosinofílica. Finalmente, la tercera y última fase es la de la sistematización por vasculitis. Esta fase de la GACS puede ser similar a la que se produce en la panarteritis nodosa.
Los territorios más afectados en la GACS son el aparato respiratorio, la piel, el sistema nervioso, el aparato gastrointestinal, el corazón y el sistema musculoesquelético. La afección pulmonar se da aproximadamente en las tres cuartas partes de los enfermos. Radiológicamente es característica la fugacidad de los infiltrados periféricos, mientras que la cavitación, las adenopatías hiliares y el patrón intersticial son más raros. La eosinofilia sanguínea, característica del síndrome, puede no ser objetivada, bien por la toma de corticoides, bien por las fluctuaciones de la cifra de eosinófilos propia de la historia natural de la enfermedad. Los lugares más accesibles y de alta rentabilidad diagnóstica son la piel, músculo estriado (en general, masa estriada) y nervio periférico, en la zona más afectada según el electromiograma. Si los resultados repetidos de esta prueba son negativos, está indicado llevar a cabo una biopsia de pulmón, riñón o de cualquier otro órgano que esté afectado. Los tres hallazgos descritos por Churg y Strauss como característicos de esta vasculitis (angeítis necrotizante, infiltración eosinofílica y granulomas extravasculares) no suelen aparecer al mismo tiempo ni en la misma pieza de biopsia.
La GACS responde habitualmente a los corticoides. Su administración produce una respuesta excelente de toda su sintomatología. Se deben mantener dosis elevadas hasta que se consigue la remisión. En ese momento, se comienza la reducción gradual de la dosis. La afectación cardíaca y renal require dosis elevadas y tratamientos más prolongados. Otros tratamientos como la azatioprina, ciclofosfamida, clorambucil y la plasmaféresis se han utilizado en pacientes con mala respuesta a esteroides o con inicio fulminante de la enfermedad.
Las concentraciones elevadas de eosinófilos periféricos pueden ser indicativas de actividad. Sin embargo, se reconoce el incremento de la proteína C reactiva y de la IgE sérica como los datos más válidos de re agudización de la vasculitis.
Granulomatosis linfomatoide
La granulomatosis linfomatoide ( GL ) fue definida en 1972 por Liebow et al. como una enfermedad granulomatosa y proliferativa angiocéntrica y angiodestructiva, y plantearon su diferenciación del linfoma pulmonar y de la granulomatosis de Wegener. Destacaron la localización extraganglionar, preferentemente pulmonar, cerebral, renal y cutánea, el curso rápido de la enfermedad y la mortalidad muy elevada, generalmente a causa de las lesiones pulmonares. Siete años después, Katzenstein et al. publicaron un estudio clínico patológico de 152 pacientes con GL donde, además de analizar el conjunto de presentaciones clínicas y de describir la frecuente afectación multiorgánica durante su evolución, recogieron un 12% de pacientes con linfomas ganglionares de alto grado en el curso de la enfermedad.
Sin embargo, en la actualidad se considera que forma parte del espectro de síndromes linfoproliferativos T4. Las lesiones angioinvasivas (LAI) pueden dividirse en tres grados histológicos que tienen implicaciones pronósticas: el grado I corresponde a lo que antes se denominaba granulomatosis linfomatoide; las lesiones del grado II son intermedias y presentan datos de atipia celular, pero no son definitivamente malignas, y el grado III corresponde a un linfoma de alto grado. Aunque la granulomatosis linfomatoide (LAI grado I) no es primariamente una vasculitis, alrededor del 50% de los pacientes presentan una remisión sostenida tras un tratamiento inmunosupresor/citotóxico, que ha demostrado ser eficaz en las vasculitis sistémicas. Se están realizando estudios para evaluar la eficacia de los protocolos de quimioterapia diseñados para los linfomas en las LAI grados I-III.
Granulomatosis Broncocéntrica
La Granulomatosis Broncocéntrica ( GBC) viene definida por la existencia de una lesión granulomatosa necrotizante, de localización exclusiva pulmonar, que afecta predominantemente a bronquios y bronquiolos, sin vasculitis (sólo incidentalmente se afectan los vasos por vecindad) y cuyo carácter broncocéntrico la diferencia fundamentalmente de los otros cuatro tipos de granulomatosis descritas por Liebow.
Los granulomas están formados por células epiteloides rodeadas por un infiltrado histiocitario y por placas de necrosis que, en un 60% de los casos, están constituidas por restos de neutrófilos y en los demás casos por detritus de eosinófilos. Otros hallazgos distintivos son la presencia de tapones de moco en los pequeños bronquiolos, la neumonía eosinofílica en los alveolos distales y, en algunos casos, la presencia de cristales de Charcott-Leyden en los granulomas.
Clásicamente se distinguen dos formas de presentación de la GBC según haya o no historia de asma, asociándose en el primer caso frecuentemente con aspergilosis broncopulmonar alérgica. En los pacientes sin historia de asma la GBC se ha descrito asociada a diversas enfermedades como dabetes, eczema, fibre del heno, artrosis, artritis reumatoide, episcleritis, vasculitis, alergias medicamentosas, EPOC, equinococosis, infecciones por micobacterias y granulomatosis de Wegener.
En general, los hallazgos radiológicos son muy variados y no se encuentran diferencias entre los pacientes asmáticos y no asmáticos. La afectación suele ser unilateral y unilobar en la mayor parte de los casos. Las imágenes pueden adoptar las caracteristícas de masa, condensación alveolar y afectación intersticial.
En la gran mayoría de los casos de GBC descritos, el diagnóstico se ha llevado a cabo por el estudio anatomopatológico de la pieza de exéresis. En cuanto al tratamiento, la extirpación del lóbulo enfermo a menudo tiene efectos curativos, aunque algunos pacientes requieren esteroides y/o citostáticos durante períodos variables de tiempo tras la cirugía. Se ha descrito la remisión espontánea de las lesiones tan sólo en un 11% de los casos.