APROXIMACIÓN AL TRATAMIENTO DE LA EPI
Dado el amplio panorama de enfermedades pulmonares intersticiales con patogenia y sustrato histológico tan dispares no se pueden dar unas normas de tratamiento generales para todas ellas. Vamos a centrarnos fundamentalmente en la FPI exponiendo al final algunas peculiaridades del manejo terapéutico de otras EPI.
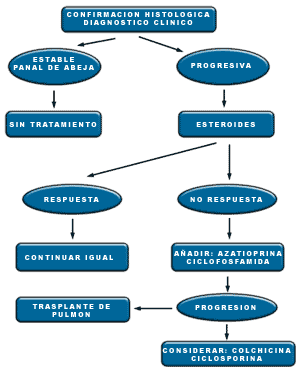
Quizá la decisión más importante es si debemos ó no tratar al paciente. El curso de la FPI es muy variable, aunque desde el momento en que está establecida la fibrosis, en general se limitan de forma muy considerable las posibilidades terapéuticas. Los fármacos más ampliamente utilizados son los esteroides, aunque no existe ningún estudio que los haya comparado con placebo para conocer su verdadera eficacia. Realmente sólo una minoría de pacientes responderán de forma objetiva (con mejoría de los parámetros funcionales respiratorios). Existen varios factores asociados a mejor pronostico y mayor tasa de respuesta a los esteroides; menor edad, sexo femenino, menor tiempo de evolución de los síntomas, linfocitosis en el BAL, biopsia pulmonar con predominio inflamatorio y fibrosis escasa y patrón en vidrio deslustrado en la TACAR.
El tratamiento inicialmente será prednisona a dosis de 0.8-1 mg/kg/día. Esta dosis se mantendrá en torno a cuatro semanas, disminuyéndola luego 5 mg cada semana hasta dejar una dosis de mantenimiento de 0.2 mg/Kg/día. No existe uniformidad en cuanto al tiempo que deben mantenerse los esteroides, pero si existe respuesta clínica quizá se pueda alargar el tratamiento 1-2 años. El seguimiento debe realizarse los tres primeros meses con periodicidad mensual y después trimestralmente, incluyendo pruebas de función pulmonar y técnicas de imagen. De éstas, la TACAR ha desplazado en gran medida a la radiografía convencional de tórax, ya que permite valorar mucho mejor la evolución de las lesiones e incluso realizar cuantificación de los cambios irreversibles, como panalización o bronquiectasias por tracción.
Además de esteroides, se han utilizado diferentes fármacos en un intento de mejorar la pobre tasa de respuesta obtenida. Quizá sean los inmunosupresores los que han producido mayores expectativas. De éstos, la ciclofosfamida y la azatioprina son los más usados. No se ha podido demostrar aumento de la supervivencia con ninguno de ellos, pero existe un trabajo realizado por el grupo de Raghu que parece indicar en un modelo de regresión múltiple potencialmente mayor supervivencia en un grupo de pacientes tratados con azatioprina/prednisona versus prednisona sola. Las dosis de azatioprina recomendadas son 3 mg/kg/día manteniendo un contaje de leucocitos por encima de 4000. En general, los efectos secundarios son menores en la terapia combinada, ya que las dosis de esteroides también son inferiores, siendo muy bien tolerada la azatioprina. La ciclofosfamida es un fármaco que plantea más problemas, sobre todo en relación con cistitis hemorrágicas, neoplasias e incluso neumonitis intersticial. La dosis habitual es de 2mg/kg/día sin sobrepasar los 200 mg/día. Existen algunos estudios en general con un número limitado de pacientes con otros fármacos antifibróticos como colchicina o D-penicilamina pero sin resultados concluyentes. Un fármaco que parece prometedor es la ciclosporina, pero se necesita más información y su uso por el momento debe restringirse a ensayos controlados.
La propuesta de tratamiento a la luz de los datos de que disponemos sería utilizar prednisona ó metil-prednisolona según la pauta previamente comentada a la que quizá se podría añadir desde el principio azatioprina, la cual va a permitir una menor dosis minimizando los efectos secundarios. En casos refractarios de evolución rápida podría probarse algún fármaco antifibrótico, preferiblemente colchicina (0.5-0.75 mg/día).
Por último, en los pacientes donde no exista contraindicación, el trasplante pulmonar es una opción que siempre hay que sopesar cuando se llega a estadios finales.
Actualmente se encuentran en fases muy precoces de investigación alternativas terapéuticas centradas en inhibidores de algunas citokinas, moléculas de adhesión ó factores de crecimiento, pero deberemos esperar un tiempo hasta que su uso clínico sea cotidiano.
En cuanto a otros tipos de EPI, siempre que exista una causa desencadenante el mejor tratamiento será evitarla, como es el caso de tóxicos, fármacos ó en neumonitis por hipersensibilidad. En situaciones concretas como vasculitis se utiliza plasmaféresis ó inmunoglobulinas intravenosas. La linfangiomiomatosis podría beneficiarse en algún caso de terapia con antiestrógenos. La sarcoidosis suele responder muy bien a los esteroides y en estadio I y es posible que en el II se puede mantener una actitud expectante. Para la mayor parte de las EPI en fase fibrótica, los fármacos a emplear son los mismos que en la FPI.